
The Strength of Taiwan’s Healthcare System Has Become the Best Defense in Combating the COVID-19 Pandemic
Author(s)
Shie-Liang HsiehBiography
Shie-Liang Hsieh is currently a distinguished research fellow in the Genomics Research Center, Academia Sinica. He has won many awards and is a National Chair Professor awarded by the Ministry of Education.
Academy/University/Organization
Academia SinicaSource
https://www.nature.com/articles/s41467-019-10360-4 https://www.frontiersin.org/articles/10.3389/fimmu.2019.02867/full-
TAGS
-
Share this article
You are free to share this article under the Attribution 4.0 International license
- LIFE SCIENCES
- Text & Image
- May 21,2020
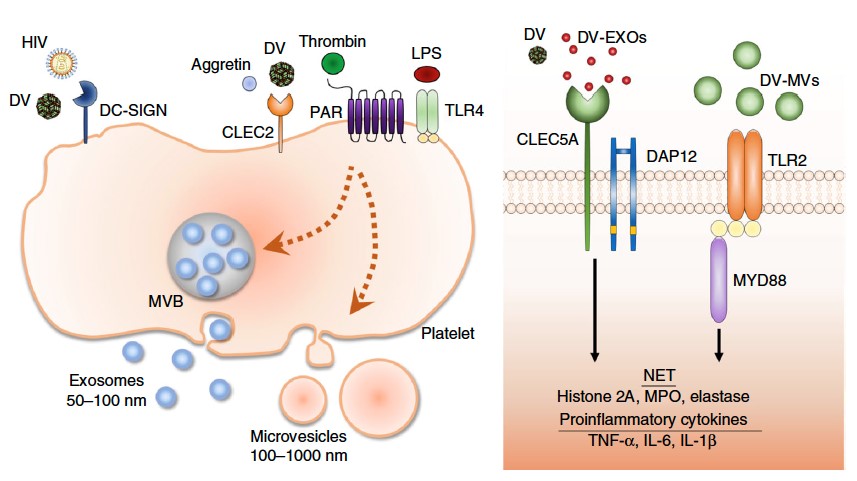
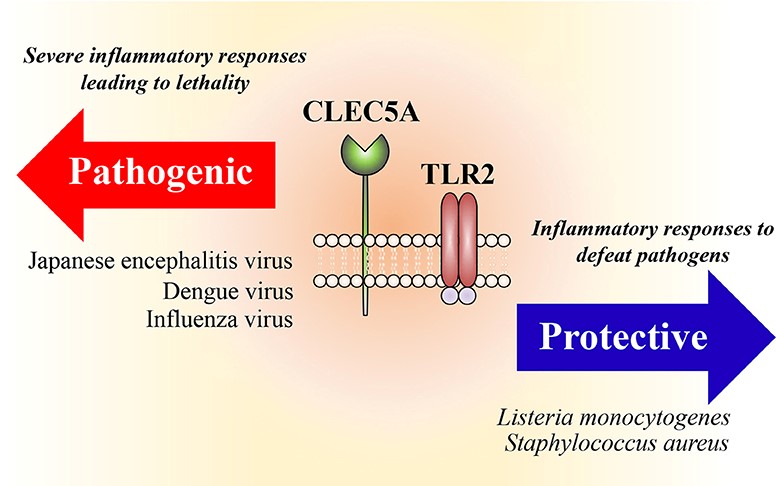
Acute viral infections frequently cause cytokine storms, which are due to massive cytokines and chemokines being released from infected cells and leukocytes. The protective roles of endosomal toll-like receptors (TLRs) and cytosolic nucleic acid sensors (RIG-I, MDA5, STING) are well elucidated, but the pathogenic host factors during viral infections remain unclear. The spleen tyrosine kinase (Syk)-coupled C-type lectins (CLECs), CLEC2 and CLEC5A, are highly expressed on platelets and myeloid cells, respectively. CLEC2 has been shown to recognize snake venom aggretin and the endogenous ligand podoplanin, and acts as a critical regulator in the development of immunothrombosis. Recently, we demonstrated that both CLEC2 and CLEC5A are critical in microbe-induced ‘neutrophil extracellular trap’ (NET) formation and proinflammatory cytokine production. Moreover, activation of CLEC2 by the dengue virus (DV) and H5N1 influenza virus (IAV) induces the release of extracellular vesicles (EVs), which further enhance NETosis and proinflammatory cytokine production via CLEC5A and Toll-like receptor 2 (TLR2). These findings not only illustrate the immunomodulatory effects of EVs during platelet-leukocyte interactions, but also demonstrate the critical roles of CLEC2 and CLEC5A in acute viral infections. Blockade of CLEC5A and TLR2 by bi-specific mAbs would be promising to inhibit virus-induced cytokine storms in the future.
The global outburst of severe acute respiratory syndrome coronavirus (SARS-CoV) infection in 2003 inspired our exploration of how a viral infection can activate a massive over-production of cytokines by the host immune system–a phenomenon known as a “cytokine storm.” This results in a severe inflammatory reaction and increases systemic vascular permeability within 2–5 days following viral exposure. The common features of SARS-CoV and other acute viral infections are the early onset of inflammatory reactions with elevated local or systemic vascular permeability, before the adaptive immune system is fully activated. These observations suggest that innate immune responses contribute significantly to the pathogenesis of acute viral infections.
The common feature of acute viral infections is the rapid onset of inflammatory reactions after exposure to the virus. Clinical symptoms range from inapparent and mild fever to severe inflammation and increased endothelial permeability in major organs. Severe acute infections in humans typically occur in response to enveloped viruses, such as dengue virus (flaviviridae), chikungunya virus (togaviridae), SARS-CoV (coronaviridae), influenza virus (orthomyxoviridae), measles virus (paramyxoviridae), rabies virus (rhabdoviridae), hanta virus (bunyaviridae), and Ebola virus and Marburg virus (filoviridae). This implies that membrane components may contribute to the severe inflammatory responses.
The protective roles of endosomal toll-like receptors (TLRs) and cytosolic nucleic acid sensors are well elucidated, but the pathogenic host factors during viral infections remain unclear. Spleen tyrosine kinase (Syk)-coupled C-type lectins (CLECs) CLEC2 and CLEC5A are highly expressed on platelets and myeloid cells, respectively. CLEC5A is responsible for DV-induced hemorrhagic fever and dengue shock syndrome, which represent the most severe responses to DV infection and are characterized by plasma leakage due to increased vascular permeability. Blockade of the DV-CLEC5A interaction suppresses the secretion of proinflammatory cytokines, but without affecting interferon-α release. Recently, CLEC2 was shown to be a novel pattern recognition receptor for DV, where DV infection activates platelets to express CD62p, CD63 and to release extracellular vesicles (EVs), including microvesicles (MVs) and exosomes (EXOs). While EXOs and MVs from resting platelets do not have any activity, DV-EXOs and DV-MVs are potent “endogenous danger signals” which trigger the activation of CLEC5A and TLR2, respectively, to promote NETosis and the production of proinflammatory cytokines in neutrophils and macrophages. While blockade of CLEC5A offers ~30% protection rate, simultaneous blockade of CLEC5A and TLR2 further increases the mice survival rate up to 90%. These observations indicate that CLEC5A/TLR2 is not a critical DV-induced pathogenesis, but also plays important roles in platelet-leukocyte interactions via recognizing platelets-derived EXOs and MVs. Thus, targeting CLEC5A/TLR2 has the potential to underpin novel strategies for treating acute viral infections.
RELATED

STAY CONNECTED. SUBSCRIBE TO OUR NEWSLETTER.
Add your information below to receive daily updates.